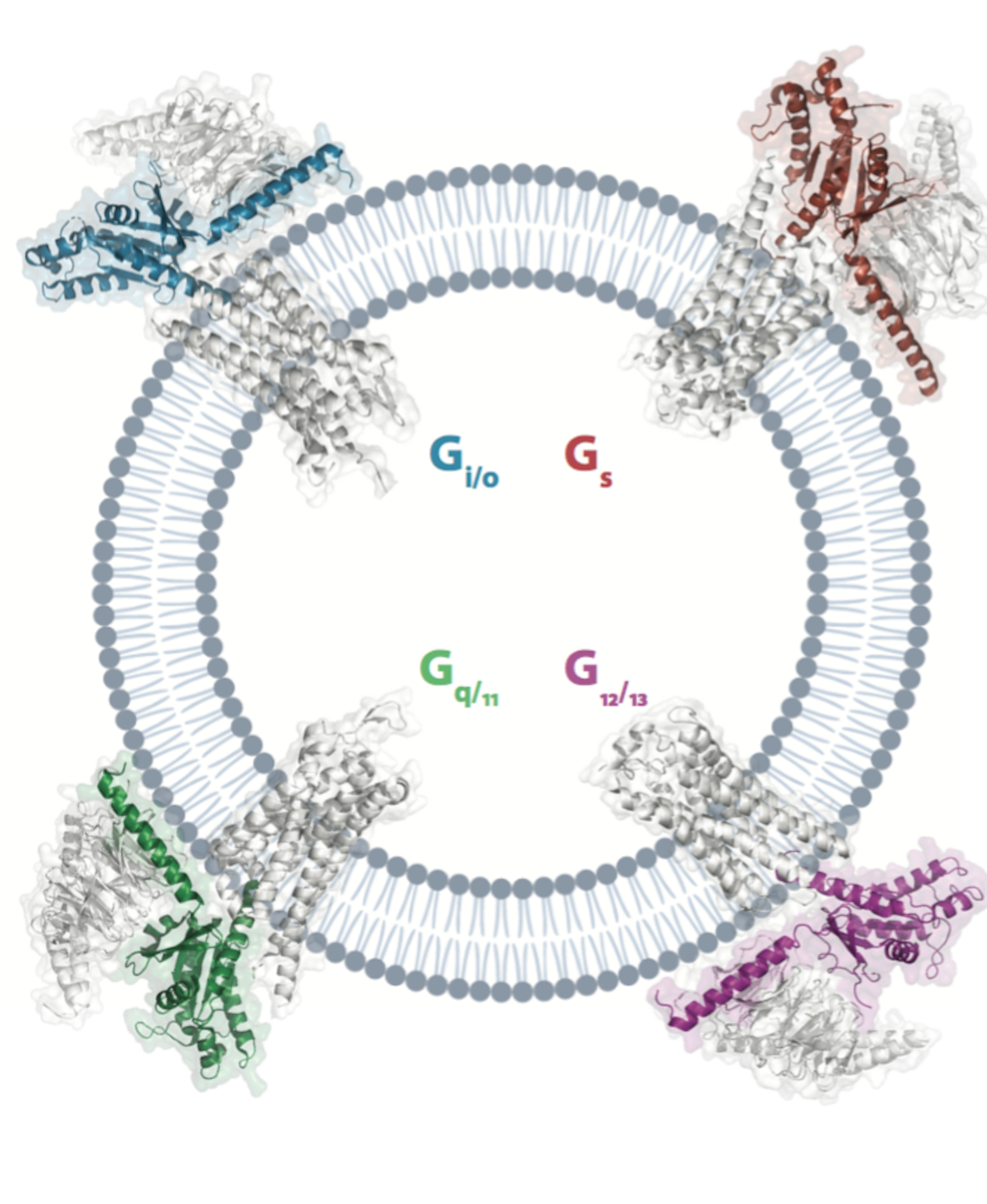
È stata effettuata dal gruppo di bioinformatica del Laboratorio di Biologia della Scuola Normale, in particolare dai perfezionandi Marin Matic e Pasquale Miglionico sotto la supervisione del professor Francesco Raimondi. Lo studio consentirà di accelerare lo sviluppo di farmaci di precisione, nonché il disegno di recettori artificiali per lo studio controllato di queste vie di segnalazione in vivo.
PISA, 28 luglio 2023. La più esaustiva indagine computazionale strutturale di complessi GPCR-proteine G mai effettuata è stata compiuta da ricercatori del Laboratorio di Biologia della Scuola Normale Superiore, in particolare dal gruppo di bioinformatica, sotto la supervisione dal professor Francesco Raimondi.
I recettori accoppiati a proteina G (GPCRs) - G protein-coupled receptors - sono la più grande famiglia di geni (poco meno di un migliaio nel genoma umano) codificanti recettori transmembrana, che mediano la trasduzione di stimoli dall’esterno all’interno della cellula mediante l’accoppiamento selettivo a proteine G, regolando molteplici aspetti della segnalazione cellulare. Il loro cattivo funzionamento è responsabile di molteplici stati patologici, da malattie neurologiche, a quelle cardiocircolatorie, al cancro. Rivestono pertanto un’enorme importanza dal punto di vista farmacologico, essendo bersagliati da circa un terzo dei farmaci approvati.
Il lavoro consentirà di accelerare lo sviluppo di farmaci di precisione, nonché il disegno di recettori artificiali per lo studio controllato di queste vie di segnalazione in vivo.
Lo studio, pubblicato su Nature Communications , è volto a comprendere i determinanti strutturali responsabili dell’accoppiamento selettivo di GPCRs a proteine G mediante l’analisi computazionale di strutture sperimentali disponibili (all’incirca 360 depositate nel Protein Data Bank a Marzo 2023), nonché predette mediante l’algoritmo di intelligenza Arficiale AlphaFold-multimer (circa 900 coppie recettori-proteine G note interagire sperimentalmente).
Con un approccio analitico guidato dai dati (data-driven), svolto dai perfezionandi Marin Matic e Pasquale Miglionico, è stato dimostrato che le caratteristiche strutturali dei complessi possono spiegare, almeno in parte, la selettività di accoppiamento. Infine, attraverso un approccio di disegno razionale computazionale, sono state suggerite mutazioni in grado di scambiare la specificità di accoppiamento, che sono state validate sperimentalmente dai collaboratori dell’Università di Tohoku, Prof. Asuka Inoue e Manae Tatsumi.
Lo studio è stato reso possibile grazie ai finanziamenti del Dipartimento di Eccellenza della Scuola Normale Superiore, dell’Associazione Italiana per la ricerca sul Cancro (AIRC, PI: Francesco Raimondi) e alle risorse di calcolo messe a disposizione dal centro HPC della Scuola e del Centro Italiano di Supercalcolo Cineca.